MDR 和 IVDR 中的 PMS:监管背景与结构
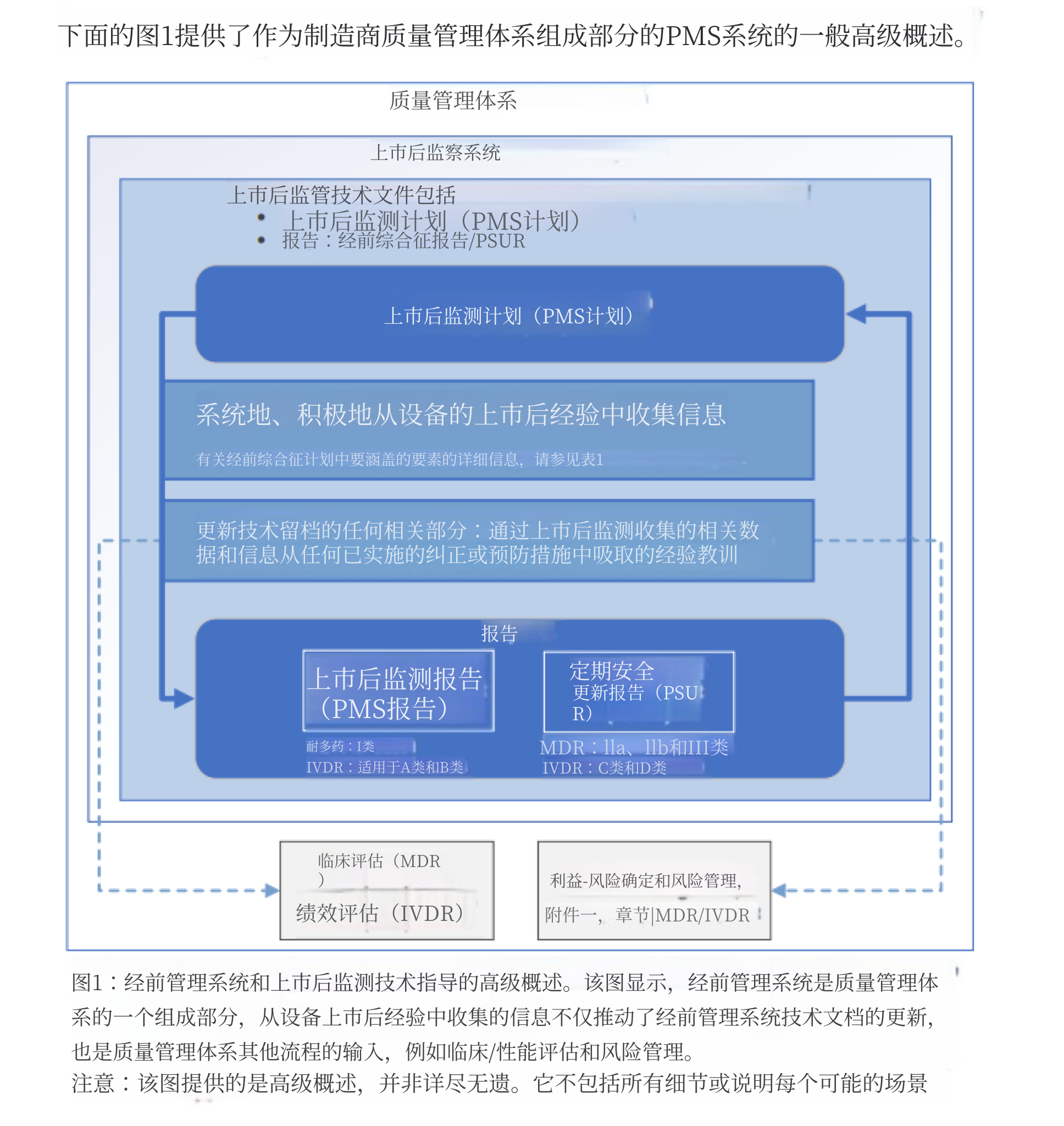
质量管理体系(QMS)、PMS 系统、包含 PMS 计划和 PMS 报告 / PSUR 的技术文件
在《欧盟医疗器械法规》(EU)2017/745(MDR)和《体外诊断医疗器械法规》(EU)2017/746(IVDR)的框架下,上市后监管(PMS)被理解为一个结构化、主动且持续的系统,是制造商质量管理体系(QMS)的组成部分。根据 MDR 第 10(10) 条和第 83 条 / IVDR 第 78 条的规定,PMS 系统旨在主动、系统地收集、记录和分析设备在整个生命周期中的质量、安全性和性能数据。
该系统建立在 PMS 计划之上(MDR 第 84 条 / IVDR 第 79 条,详见两部法规的附件 III),该计划定义了将收集哪些信息、信息来源以及收集方法。上市后监管的技术文件是整体技术文件的一个明确子集,主要包括 PMS 计划和 PMS 输出,即 PMS 报告和/或定期安全更新报告(PSUR),具体取决于器械类别和监管框架。该系统的输出通过 PMS 报告(MDR 第 85 条 / IVDR 第 80 条,适用于 I 类器械和 A、B 类 IVD)或定期安全更新报告(PSUR)(MDR 第 86 条 / IVDR 第 81 条,适用于 IIa、IIb 和 III 类器械以及 C、D 类 IVD)形式化呈现。
正如 MDCG 2025-10 的高层概览所示,PMS 计划推动了上市后经验的持续收集,这些经验反过来被纳入技术文件,并直接与其他关键 QMS 流程(如风险管理和效益–风险评估〔MDR/IVDR 附件 I〕以及临床或性能评价)相互衔接,确保真实世界数据系统性地支持监管合规和生命周期决策。
核心信息
PMS 必须是结构化、主动且贯穿整个生命周期的
MDCG 2025-10 重申了 MDR/IVDR 的核心原则:PMS 并不是被动的投诉处理活动,而是一个贯穿器械整个生命周期的持续、结构化和主动的系统,从首次投放市场/投入使用开始,一直到最后一台设备预期寿命的结束。同时强调,上市后监管的规划应当在器械开发阶段就已经开始,以确定在器械进入市场后将开展哪些活动来系统性地收集其性能信息。
两个重要启示:
- “主动”不是可选语言。 指南明确指出,主动的 PMS 是有意地寻求超越投诉渠道的信息,例如文献筛查、用户反馈、注册/登记(在适用情况下)、客户调查、PMCF/PMPF 等。
- PMS 输出必须驱动决策。 结论及后续行动需要通过 PMS 报告或 PSUR 进行记录,而下一轮 PMS 循环可能需要根据所获得的经验修订 PMS 计划。
MDCG 2025-10 的实用价值
一个可以映射到 QMS 的 “PMS 运行模型”
指南将 PMS 描述为一个实际的循环:
信息来源 → 收集 → 评估/分析 → 结论 → 行动 → 更新 QMS 与技术文件
这不仅是概念性的。文件强调 PMS 信息会持续用于更新关键的 QMS 要素,尤其是风险管理、效益–风险评估以及临床/性能评价。
换句话说,审计员可能会问类似的问题: “请展示 PMS 信号如何触发风险管理文件、临床评价报告、标签/说明书 (IFU) 以及 CAPA 决策的更新。”
“主动数据收集”被进一步强化
(这提高了 PMS 计划的要求)
MDCG 2025-10 强调 PMS 计划是整个系统的引擎。它重申计划必须定义监测内容、频率以及方法,并根据风险等级、器械类型和真实使用场景来选择。表 1 特别有用,简明总结了 PMS 计划必须涵盖的要素(附件 III 第 1 节)。
审计员可能会关注的计划要素包括:
- 明确的指标与阈值,用于持续重新评估效益–风险与风险管理(不仅仅是“我们统计投诉”)。
- 趋势报告准备度:用于检测事件频率/严重性显著增加的统计方法与观察期。
- 投诉调查方法:与器械风险相匹配的有效工具/方法,尤其是高风险产品组合。
- 可比/“类似产品”情报:作为系统化的输入,结合最新技术(SOTA)监测,而不是非正式的市场传闻。
- 一个细微但重要的点:指南建议 PMS 计划可以定义“使用哪些方法”,而具体的“如何/由谁执行”可以放在引用的 SOP 中,只要计划保持具体且可追溯。
数据质量的重要性
指南明确警告弱数据来源。它特别提到不可验证的数据(甚至包括公共/社交媒体)可能导致过度反应,并提醒制造商在分析前必须考虑数据质量与完整性。
这并不意味着要忽视“嘈杂”渠道,而是要记录:
- 如何筛选这些输入,
- 如何验证或决定不验证,
- 如何防止它们扭曲趋势分析或 CAPA 启动。
定制医疗器械 (CMDs):PMS 要求被明确强化
MDCG 2025-10 专门说明定制医疗器械并不豁免于 MDR 的 PMS 要求。制造商仍需建立 PMS 系统,并规划/记录生产后的经验,包括 PMCF,并通过分组(相同用途/材料/工艺/设计原则)来管理,而不是将每个 CMD 独立作为完整生命周期文件。
CMD 制造商必须:
- 为 I 类 CMD 提供 PMS 报告,
- 为 IIa/IIb/III 类 CMD 提供 PSUR, 并将这些文件纳入 CMD 文档要求。
制造商的重点行动方向
若要快速、可辩护地与 MDCG 2025-10 对齐,以下是最高回报的措施:
确保 PMS 计划涵盖:主动信息来源、分析方法、指标/阈值、投诉调查方法、趋势报告方法与观察期、沟通协议(CA/NB/经济运营商/用户)、以及纠正措施范围的可追溯工具。
指南明确指出:PMS 发现必须持续反馈到效益–风险与风险管理,以及临床/性能评价。如果 PMS 识别出新的副作用或缺陷,风险管理流程必须跟进。 一个实用的证据方式是维护简单的“信号到更新”追踪: PMS 信号 → 评估记录 → 决策(无行动 / CAPA / FSCA / 标签更改 / CER 更新) → 更新文件引用。
- 在 PMS 报告/PSUR 中清晰呈现“结论 + 行动”
文件强调结论和后续行动必须记录在 PMS 报告或 PSUR 中,并且 PMS 计划可能需要根据循环结果进行修订。审计员尤其喜欢看到这个闭环。
这不是新法律,但会塑造监管预期
MDCG 指南并不像法规那样具有法律约束力(文件本身也包含标准免责声明),但它强烈影响主管当局和公告机构对“良好实践”的解读。
因此,即使你已经在做 PMS,问题也变成:你的 PMS 系统是否符合 MDCG 2025-10 所描述的主动、基于风险、与 QMS 深度整合的模型?